December 2, 2021

The following article was originally contributed to and published within the surfaces.org’s “SurFACTS in Biomaterials” Fall 2021 Volume 26, Issue 3 newsletter. View their newsletter archive here.

Medical devices make up a substantial segment of our healthcare operations for the diagnosis and treatment of health conditions. The creation and approval of new devices is important to the continued improvement of the overall health, improved diagnosis, and availability of treatment options for patients.
Section 201(h) of the Food, Drug and Cosmetics Act defines a medical device as “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory…which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes.” This includes implantable devices and devices designed to deliver a drug or biologic to a patient. In the latter case, the drug or biologic being delivered is usually evaluated separately by the FDA Center for Drug Evaluation and Research (CDER) or the Center for Biologics Evaluation and Research (CBER), while the FDA Center for Devices and Radiological Health (CDRH) evaluates medical devices.

 Unlike requirements for investigative drug/biologics products, only a small percentage of devices require clinical data to demonstrate they are both safe and effective prior to market approval. The FDA classifies medical devices into Class I, II, and III. Most medical devices can be classified by finding the matching description of the device in Title 21 of the Code of Federal Regulations (CFR), Parts 862-892. For each of the devices classified by the FDA the CFR gives a general description including the intended use, the class to which the device belongs (i.e., Class I, II, or III), and information about marketing requirements. Most low-risk devices may be marketed without prior FDA review, and most medium-risk devices must only determine substantial equivalence to an existing device. However, certain Class II devices and all Class III devices which pose a significant risk of illness or injury must go through the FDA’s Premarket Approval (PMA) process to evaluate safety and effectiveness.
Unlike requirements for investigative drug/biologics products, only a small percentage of devices require clinical data to demonstrate they are both safe and effective prior to market approval. The FDA classifies medical devices into Class I, II, and III. Most medical devices can be classified by finding the matching description of the device in Title 21 of the Code of Federal Regulations (CFR), Parts 862-892. For each of the devices classified by the FDA the CFR gives a general description including the intended use, the class to which the device belongs (i.e., Class I, II, or III), and information about marketing requirements. Most low-risk devices may be marketed without prior FDA review, and most medium-risk devices must only determine substantial equivalence to an existing device. However, certain Class II devices and all Class III devices which pose a significant risk of illness or injury must go through the FDA’s Premarket Approval (PMA) process to evaluate safety and effectiveness.
The PMA process requires manufacturers submit clinical data assuring the safety and effectiveness of a device. This process is similar to (although typically less robust than) the clinical trial process for investigative drugs/biologics.
Medical device clinical studies are usually categorized as feasibility studies or as pivotal studies. A feasibility study may provide support for a future pivotal study or may be used to answer basic research questions. These studies are not intended to be the primary support for a marketing application. In feasibility studies, endpoints and sample size are generally not statistically driven, and studies average 10-40 patients. Pivotal medical device studies, however, are intended as the primary clinical support for a marketing application and must be designed to demonstrate a “reasonable assurance of safety and effectiveness.”

Medical device trials also have the potential to encounter a number of challenges that are unique from drug/biologic studies:
- Blinded, randomized, controlled trials (RCTs) – the gold standard design for drug trials – are rare within medical devices trials, as device trials are often very difficult to blind.
- Developing sham devices and procedures is complicated, and in certain scenarios may be considered unethical. However, mounting evidence has shown that medical procedures can create a strong placebo effect, and sometimes a comparative placebo arm is required by the FDA for approval.
- Resulting data depend heavily on physician technique. This amplifies the importance of proper, thorough, and timely investigator/site training along with strategic selection to ensure physicians have the right guidance, tools, and qualifications to use investigative devices in the way device companies intended them to be used.
- Device studies often include highly diverse endpoints.
- Modifications may be made to an investigational device during a clinical trial.
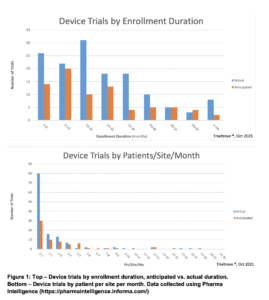
The data displayed in Figure 1, collected from Pharma Intelligence Informa, show averages of enrollment duration and the average patients/site/month rate for device trials.
As with clinical trials focused on novel drugs and biologics, low or slow enrollment may occur if sites are not selected strategically and properly managed. Site feasibility assessments and a strategic site selection process can help predict speed of enrollment and how data flow will occur over time, considering areas such as past performance, site qualifications/experience, access to patient population, and operational excellence.
RELATED RESOURCES